Teaching in courses:
Research Group
T-Side

Group leader: Vasileios Bekiaris
T cell Signaling and Development
The T-SIDE (T cell SIgnaling and DEvelopment) group focuses on cellular and molecular immunology, with a particular interest in pathways that regulate T cell development and function.
T lymphocytes are cells of our immune system that are necessary to protect from infections. Since they need to tackle many different pathogens, there are different types of T cells. Each T cell type is dedicated to a specific group of pathogens and secretes effector molecules (cytokines) evolved to fight such pathogens (Figure 1). In addition to their anti-pathogenic role, T cells protect us from cancer, while failure to dampen their activity may result in chronic inflammation or autoimmunity (Figure 1).
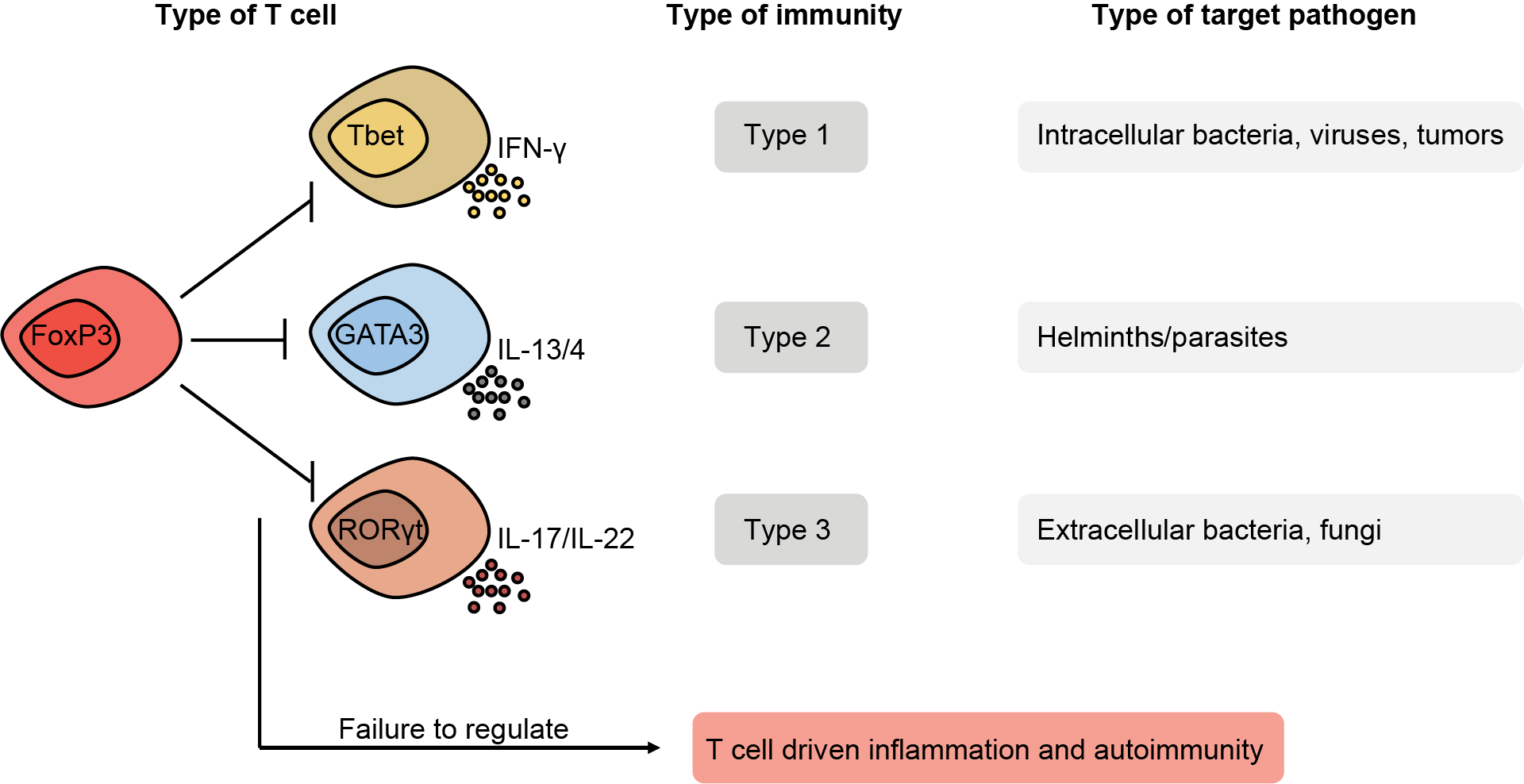
Our group is interested in T cells that produce the cytokine interleukin(IL)-17 and are termed either type-17 or type-3. IL-17-producing T cells are required to protect from infections like fungi and extracellular bacteria but have been shown to be pathogenic in many chronic inflammatory diseases such as multiple sclerosis or psoriasis (Figure 2). Understanding how these T cells develop and function will give us a tool in manipulating them for therapeutic purposes. We are particularly interested in CD4+ T helper-17 (Th17) and gamma delta (γδ) T-17 (γδT17) cells.
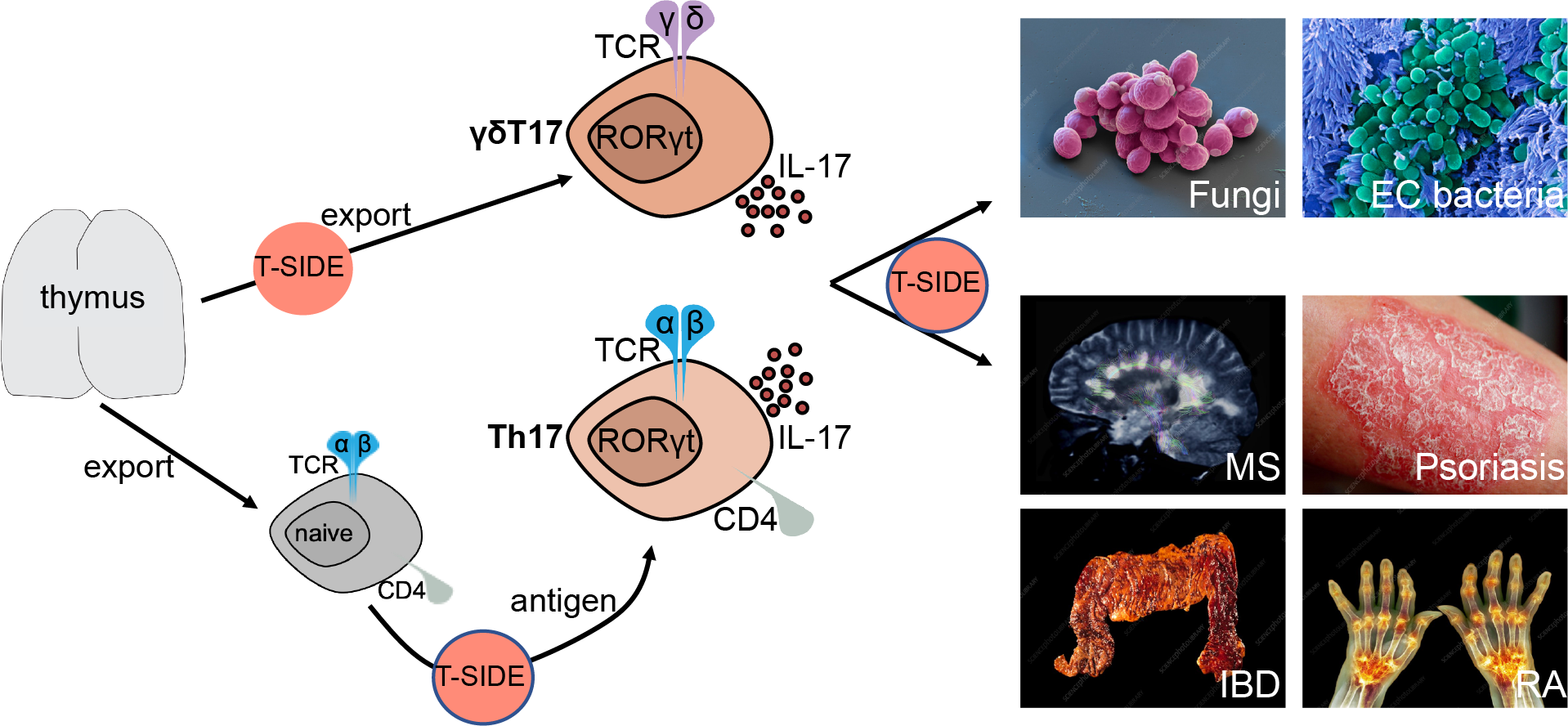
Th17 cells are part of the adaptive immune system, and differentiate in response to antigenic stimulation after their naive counterparts leave the thymus – the organ where all T cells develop. In contrast, γδT17 cells are innate – i.e. they do not require TCR-antigen interactions to respond – and acquire most of their effector functions before they leave thymus (Figure 2). At steady-state, both Th17 and γδT17 cells are found mainly at mucosal surfaces, the skin and secondary lymphoid tissues (e.g. lymph nodes), however, during infection and inflammation they can migrate to any organ. The objective of our lab is to elucidate which signaling pathways and molecules regulate the development, differentiation and function of Th17 and γδT17 cells (Figure 2).
Projects
Ongoing research projects
Checkpoint inhibition of γδ T cells in cancer
Irrespective of our socio-economic or genetic background, there is a 40% chance that we will deal with cancer at some point in our lives. This is a grim statistic and one of the reasons why cancer is a leading cause of mortality worldwide. T cells are necessary to fight malignant cells and cancer. However, the tumor microenvironment (TME) has the ability to suppress T cell function. This is achieved by induction of a group of molecules on the surface of the T cell, which are collectively known as immune checkpoint receptors (ICRs). These receptors are normally upregulated during all T cell responses and their role is to transmit a negative stop signal in order to prevent overactivation and subsequent autoimmunity. However, they are expressed only transiently and thus the T cell can respond to further challenges. In marked contrast, the TME induces constitutive ICR expression on T cells, rendering almost permanently inactive, and therefore unable to fight the growing tumor cells. Blockade of ICRs with specific antibodies can decelerate the negative signal, restore T cell functionality and allow a productive anti-tumor response (Figure 1). This technique is widely used in the clinic for certain cancers and is collectively known as checkpoint inhibitor (CPI) therapy.

Figure 1. Overview of the mechanism by which CPI therapy works in cancer. (A) Under a normal response T cells receive positive signals (e.g. from their TCR) to become activated. Transient upregulation of ICRs prevents T cell overactivation. (B) In cancer ICRs remain constitutively expressed leading to unresponsive T cells and subsequent tumor escape. (C) CPIs block ICRs and render T cells responsive again, which can now fight the cancer.
There are numerous demonstrations that activated IFN-γ-producing cytotoxic γδ T cells have a protective role against different kinds of cancer. However, there is also increasing evidence that IL-17-producing γδ T cells (γδT17) promote tumor growth (Figure 2).

Figure 2. Anti-tumor and pro-tumor γδ T cells. Subsets of γδ T cells that produce the cytokines TNF and IFN-γ and have cytolytic activity through the production of perforin and granzymes (Gzms) are potent anti-tumor effectors. However, γδ T cell subsets that produce the cytokine IL-17 often favor tumor growth.
CPI therapy will potentially target both anti-tumor and pro-tumor γδ T cells, with the latter case leading to poor clinical outcome for the patient. In this regard, we have shown that γδT17 cells are responsive to ICR signaling and can express a number of ICRs on their surface (see https://doi.org/10.1016/j.immuni.2013.10.017 and https://doi.org/10.1002/eji.201948456). It is possible that we can identify combinations of ICRs that can be targeted by CPI only in anti-tumor but not in pro-tumor γδT17 cells, accelerating therefore cancer clearance (Figure 3).

Figure 3. Manipulation of CPI therapy to improve anti-cancer γδ T responses. (A) The tumor microenvironment is poised to induce ICRs in both anti- and pro-tumor γδ T cells. Blocking ICRs expressed in both subsets will release an anti-tumor response but also pathogenic pro-tumor events. (B) If different ICRs are expressed in the different γδ T cell subsets then we can design CPI therapies that target only the anti-tumor response, thus avoiding pathogenicity from the pro-tumor γδ subsets.
Our aim is to characterize phenotypically and transcriptionally γδ T cells from cancer patients before, during and after CPI therapy in order to correlate successful clinical outcome with γδ T cell functionality and to pinpoint ICRs or groups of ICRs that might be differentially specific for anti-tumor versus pro-tumor populations. This project is funded by the Danish Cancer Society.
cIAPs as molecular determinants of T cell function and development
The cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) are E3 ubiquitin ligases that regulate the canonical nuclear factor kappa B (NFκB) pathway downstream of tumor necrosis factor receptor 1 (TNFR1). When cIAP1/2 is abrogated, TNFR1 can lead to cell death (Figure 1). Independent of receptor-ligand interactions however, cIAP1/2 ubiquitously and constitutively repress activation of the non-canonical NFκB pathway (Figure 1). Blockade of cIAP1/2 or receptor-ligand driven displacement can activate the non-canonical NFκB pathway in any given cell (Figure 1).

Figure 1. cIAP1/2 are NFκB molecular switches. Downstream of TNFR1 cIAP1/2 are necessary for the activation of the canonical NFκB pathway and when inhibited programmed cell death is initiated. Simultaneously, cIAP1/2 block activation of the non-canonical NFκB pathway. When cIAP1/2 are inhibited or when displaced by receptor ligand interactions, the non-canonical NFκB pathway is activated.
We have shown that when Th17 cells are acutely depleted of cIAP1/2 by the small molecule inhibitor smac mimetic (SM), their transcriptional network is rewired, they lose their capacity to produce IL-17 and to induce inflammation. This was due to aberrant non-canonical NFκB activation, which simultaneously drove Th2 differentiation (Figure 2). Our data is suggestive that cIAP1/2 are necessary for Th17 differentiation and may regulate the balance between type 3 and type 2 immunity.

Figure 2. The cIAP1/2-non-canonical NFκB signaling axis regulates Th17 differentiation. Acute degradation of cIAP1/2 with the small molecule inhibitor smac mimetic (SM) during Th17 cell differentiation suppresses IL-17 production, and reduces their capacity to induce inflammation. In addition, SM-induced cIAP1/2 degradation promotes Th2 differentiation at the expense of Th17 cells. The effects of SM in Th17 were driven by activation ofnon-canonical NFκB. See https://stke.sciencemag.org/content/12/596/eaaw3469
In a follow up study, we found that SM-driven cIAP1/2 degradation leads to increased production of a number of adhesion and cytoskeleton-associated molecules. This resulted in homotypic T-T interactions that were dependent on the non-canonical NFκB pathway and integrin-mediated signaling. Moreover, in collaboration with DTU Health Tech Professor, Martin Dufva, we can demonstrate that SM-treated T cells have increased adherent capacity. This study will be published soon. In collaboration with Professor Dufva we will additionally explore methods to test whether such increased adherent capacity allows T cells to migrate better within the inflamed or tumor microenvironment, and whether they have increased killing capacity.
In addition to acute cIAP1/2 depletion we have generated a genetic mouse model whereby the gene coding for cIAP2 is knocked out in all cells, whereas the gene coding for cIAP1 is deleted only in RORγt-expressing populations. Using these mice, we have been able to show that γδT17 cells require cIAP1/2 during late neonatal life in order to maintain their transcriptional integrity and expand within lymph nodes and barrier tissues. We additionally show that lack of cIAP1/2 renders γδT17 cells unresponsive to inflammatory stimuli in the skin, whereas in the gut cIAP1/2 is important to support protection from the attaching and effacing bacterium Citrobacter rodentium (Figure 3). This study is currently in progress.
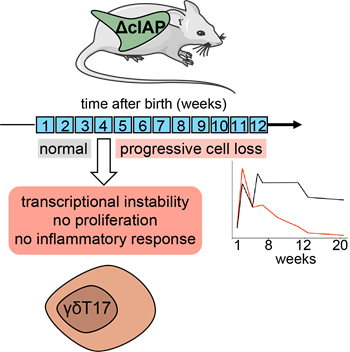
Figure 3. cIAP1/2 are necessary for γδT17 lineage stability and homeostatic turnover. Mice with global cIAP2 deficiency and RORγt-driven deletion of cIAP1 (ΔcIAP) display normal a γδT17 compartment during the first 3 weeks of life. At around week 4 expression of the transcription factors RORγt and cMAF is downregulated, the cells fail to proliferate following cytokine activation and cannot mount an inflammatory response. This leads to a progressive loss of γδT17 cells from all organs. See https://www.biorxiv.org/content/10.1101/2020.05.14.094334v1
We have recently secured funding from the Leo Foundation to use the aforementioned mice in order to test the role of cIAP1/2 in Th17 responses during psoriasis. As part of this project we will also explore the therapeutic potential of SM treatment during psoriasis and its effects on human T cells and keratinocytes.
Development and function of RORγt+Tbet+ intestinal γδT17 cells
A characteristic of Th17 cells is their high plasticity – i.e. their ability to acquire non-type 3 functions, such as production of IFN-γ. Thus, during an immune response Th17 cells can acquire expression of Tbet and co-produce IL-17 and IFN-γ, which renders them highly protective against certain infections or highly pathogenic during inflammation. Similarly, it has been shown that during infection or inflammation, but not at steady-state γδ T cells can co-produce IL-17 and IFN-γ.
During thymic development, innate-like γδ T cells can receive a strong TCR signal and differentiate into IFN-γ-producers, or they can receive weak TCR signals and become IL-17-producing. The two pathways are mutually exclusive with one antagonizing the other, especially when lineage transcription factors, such as RORγt, start to be expressed. In this regard, at steady-state there can be cells that produce IFN-γ and express Tbet or cells that produce IL-17 and express RORγt, but not both (Figure 1). Our recent data have challenged this paradigm. We discovered that RORγt-expressing γδ T cells in the small intestinal and colonic lamina propria (sLP and cLP) additionally express Tbet (Figure 1). Upon cytokine activation they could co-produce IL-17, IFN-γ and IL-22, demonstrating for the first time γδ T cell plasticity at steady-state. Intriguingly, expression of Tbet and acquisition of type 1 function occurred during the first week of life independent of microbiota, TLR or IL-12 signaling. Our data provided some evidence that Tbet expression can be regulated by STAT5. Similar to γδT17 cells in the LN, the turnover of the intestinal population depended strictly on STAT5 (Figure 1).

Figure 1. Unique characteristics of intestinal γδT17 cells. At steady-state,in the lymph node there can be either Tbet+IFN-γ+ or RORγt+IL-17+ γδ T cells. However, in the small intestinal (or colonic) lamina propria we found that most RORγt+ γδ T cells co-expressed Tbet and co-produced IFN-γ (regulated by retinoic acid receptor; RAR) and IL-17 (regulated by STAT3 along with IL-22). Induction of Tbet occurred during the first week of life and was independent of TLRs, IL-12 and the microbiota but partially dependent on STAT5. As in the lymph node, STAT5 was necessary for their neonatal turnover. See https://www.jci.org/articles/view/131241
We have generated a novel reporter mouse strain with which we can fate-map the transition of RORγt+ into Tbet+ γδ T cells. We are currently using this mouse model to investigate γδ T cell plasticity at steady-state in the gut and during systemic inflammation. Moreover, we are in the process of generating mice with RORγt-driven Tbet deletion in order to test the importance of Tbet in intestinal γδT17 cells. At the same time, with a grant funder by the Novo Nordisk Foundation we will be working to dissect the exact spatiotemporal requirements that occur during embryonic and neonatal life and lead to the development of RORγt+Tbet+ intestinal γδT17 cells.
We have recently performed RNA-seq experiments from sorted Tbet+ and RORγt+ γδ T cell populations from lymph nodes and gut and have identified many genes and pathways that are unique to the intestinal population. We are in the process of experimentally addressing the relevance of some of these pathways.
The transcriptional regulators STAT3 and STAT5 in type 3 T cell development and function
The signal transducer of activation and transcription (STAT) proteins (STAT1 through STAT6) are downstream of Janus kinase (JAK)-interacting cytokine or growth factor receptors. JAK-driven phosphorylation of STATs directs them to the nucleus where they function as transcription factors and chromatin modifiers. The JAK-STAT pathway is at the core of a lymphocyte’s life, from development all the way to effector function. In particular STAT3 and STAT5 are master regulators of T cell generation, differentiation and cytokine responsiveness, while their common disease-associated mutations highlight their importance in human biology (Figure 1). Homologues of STAT3 and STAT5 pre-date vertebrate evolution, and have shaped modern immunity in most organisms.

Figure 1. The JAK/STAT pathway. The JAK/STAT pathway is activated downstream of many cytokine and growth factor receptors and is fundamental for the immune system. STAT proteins, upon JAK-driven phosphorylation, translocate to the nucleus where they function as transcription factors and chromatin modifiers to induce or suppress genes related to numerous immune cell properties.
We have recently discovered that STAT5 is necessary for the post-thymic neonatal development of γδT17 cells by regulating early life expansion and survival within the lymph nodes and the intestine. We additionally dissected the role of the two STAT5 gene products, STAT5A and STAT5B, and showed that whereas STAT5A favors γδT17 cells, STAT5B favors IFN-γ-producing γδ T cell populations. Furthermore, mice with selective STAT5 deficiency in IL-17-producing lymphocytes where resistant to experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (Figure 2). As part of this work we described and characterized for the first time a novel gut-specific γδT17 subset that co-expresses RORγt and Tbet and co-produces IL-17 and IFN-γ. Interestingly, expression of Tbet appeared to be dependent on STAT5B (Figure 2). This work was completed in close collaboration with Professor Richard Moriggl at the University of Veterinary Medicine Vienna.

Figure 2. The neonatal microenvironment programs innate γδ T cells through the transcription factor STAT5. During neonatal life STAT5 sustains turnover of γδT17 populations in lymph nodes and intestine. Of the STAT5 proteins, STAT5A promotes γδT17 cells whereas STAT5B differentially impacts Tbet-expressing γδ T cells and regulates Tbet expression in the gut. In the absence of STAT5, adult mice have severely reduced γδT17 numbers, which results in resistance to EAE. In the gut, cytokine production RORγt and Tbet double-expressing γδT17 cells is additionally regulated by STAT3 and retinoic acid receptor (RAR). See https://www.jci.org/articles/view/131241
We are currently investigating: 1) the differential role of STAT5A and STAT5B in γδ T cell development using gain-of-function mutant mice, 2) the importance of STAT5 in regulating Th17 responses in the intestine, 3) the importance of STAT5 gain-of-function mutations in driving γδ T cell lymphomas (this project is headed by Professor Richard Moriggl in Vienna).
In contrast to STAT5, STAT3 is not required for γδT17 cell development or for their ability to produce IL-17 at steady-state. This is unlike Th17 cells, which need the IL-6-STAT3 and IL-23-STAT3 signaling axes in order to fully differentiate. However, we have shown that STAT3 is necessary for γδT17 cells to respond to inflammatory stimuli in the skin. As such, cells with RORγt-driven STAT3 deletion fail to proliferate and produce IL-22 and cannot increase IL-17 levels in response to psoriasis-like disease. We are currently collaborating with Professor Richard Moriggl at the University of Veterinary Medicine Vienna to address the role of STAT3 gain-of-function mutations in driving systemic γδ T cell responses and γδ T cell lymphomas.

Figure 3. STAT3 is critical of for γδT17 cell responses and skin inflammation. The transcription factor STAT3 is necessary to induce γδT17 cell expansion and cytokine production during skin inflammation, while STAT4 acts to sustain optimal levels of the cytokine IL-17F. Expression of STAT3 by γδT17 cells is necessary to promote psoriasis‐like inflammatory disease in mice. See https://www.embopress.org/doi/full/10.15252/embr.201948647
Group Leader
Vasileios Bekiaris Groupleader, Associate Professor Department of Health Technology Phone: +45 35886828 Mobile: 9351 1297 vasbek@dtu.dk

